Introduction to pyCyto¶
pyCyto is a Python workflow package for reproducible analysis of microscopic cytotoxicity assays. It processes time-lapse fluorescence microscopy data through a modular pipeline — from raw images to quantified cell interactions — and can run on a single workstation or scale across HPC clusters via SLURM.
Live Preview¶
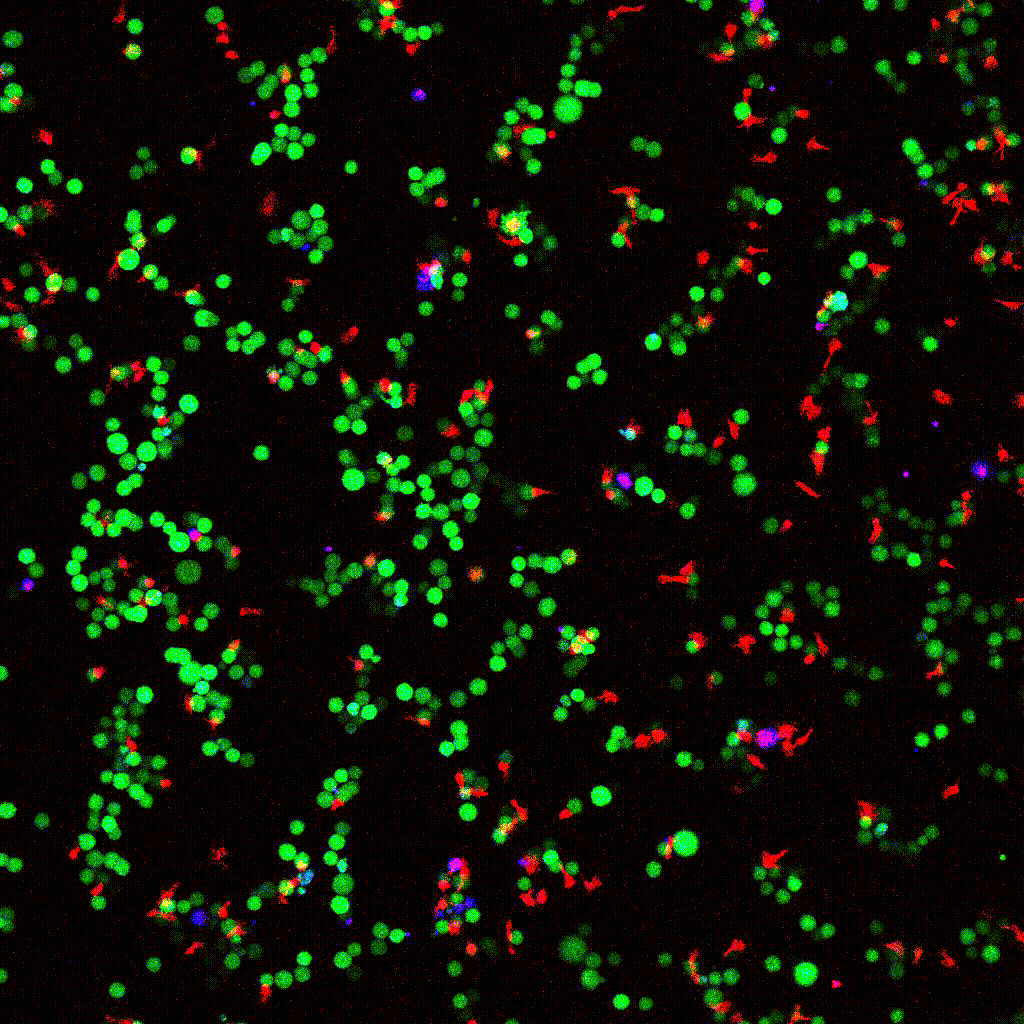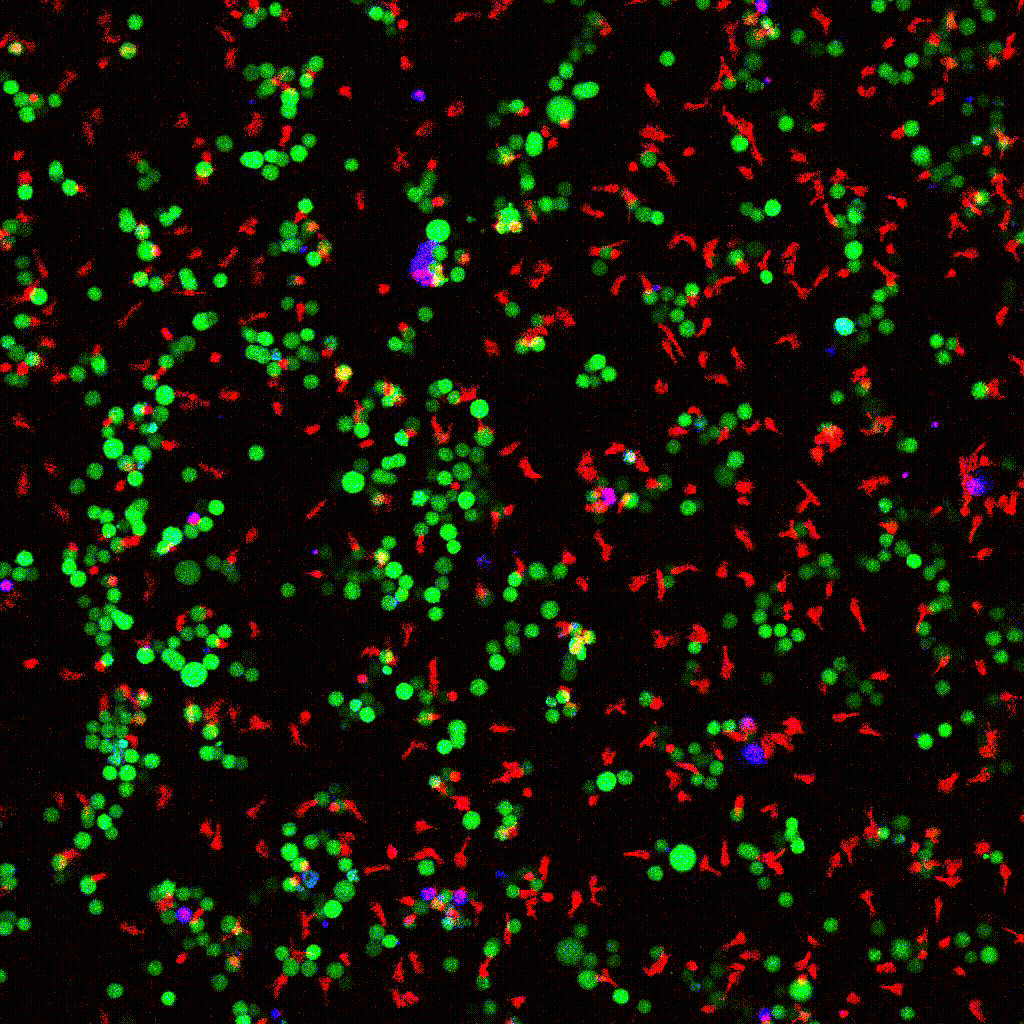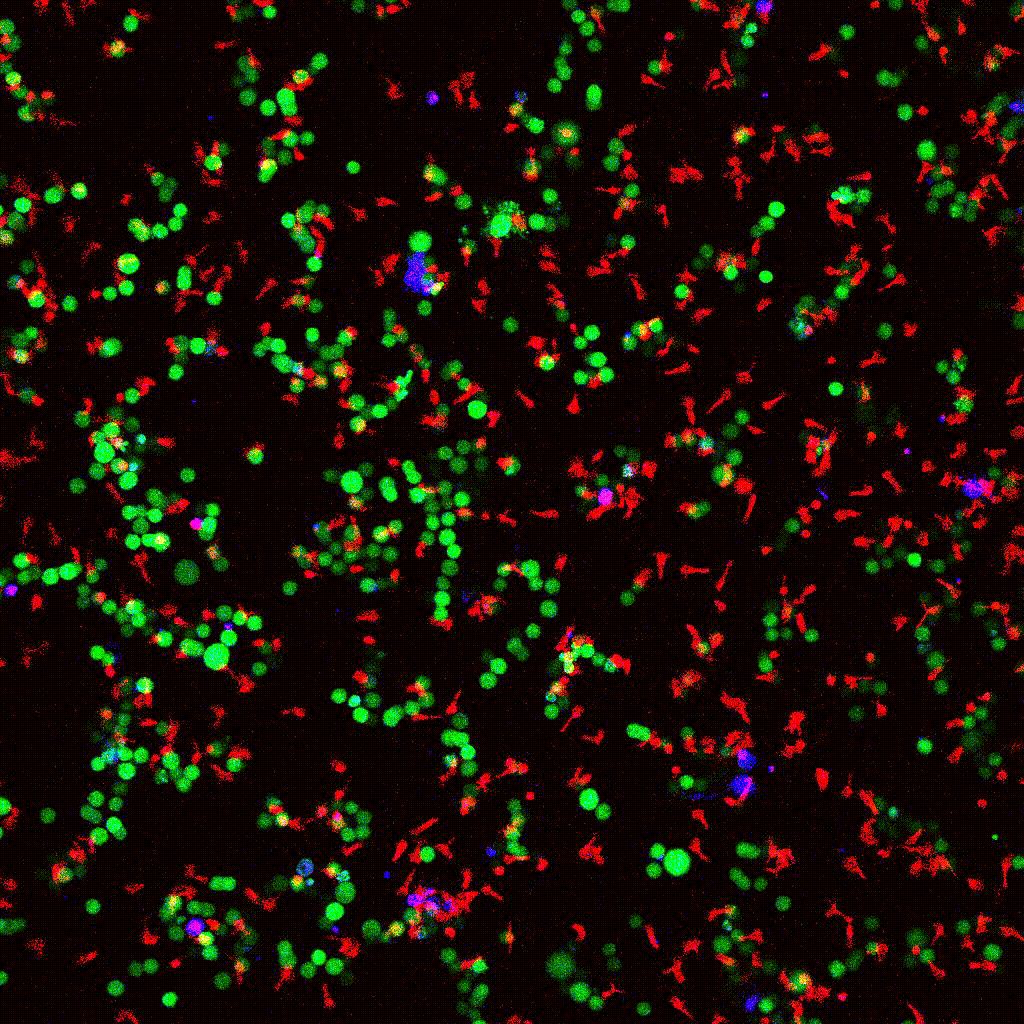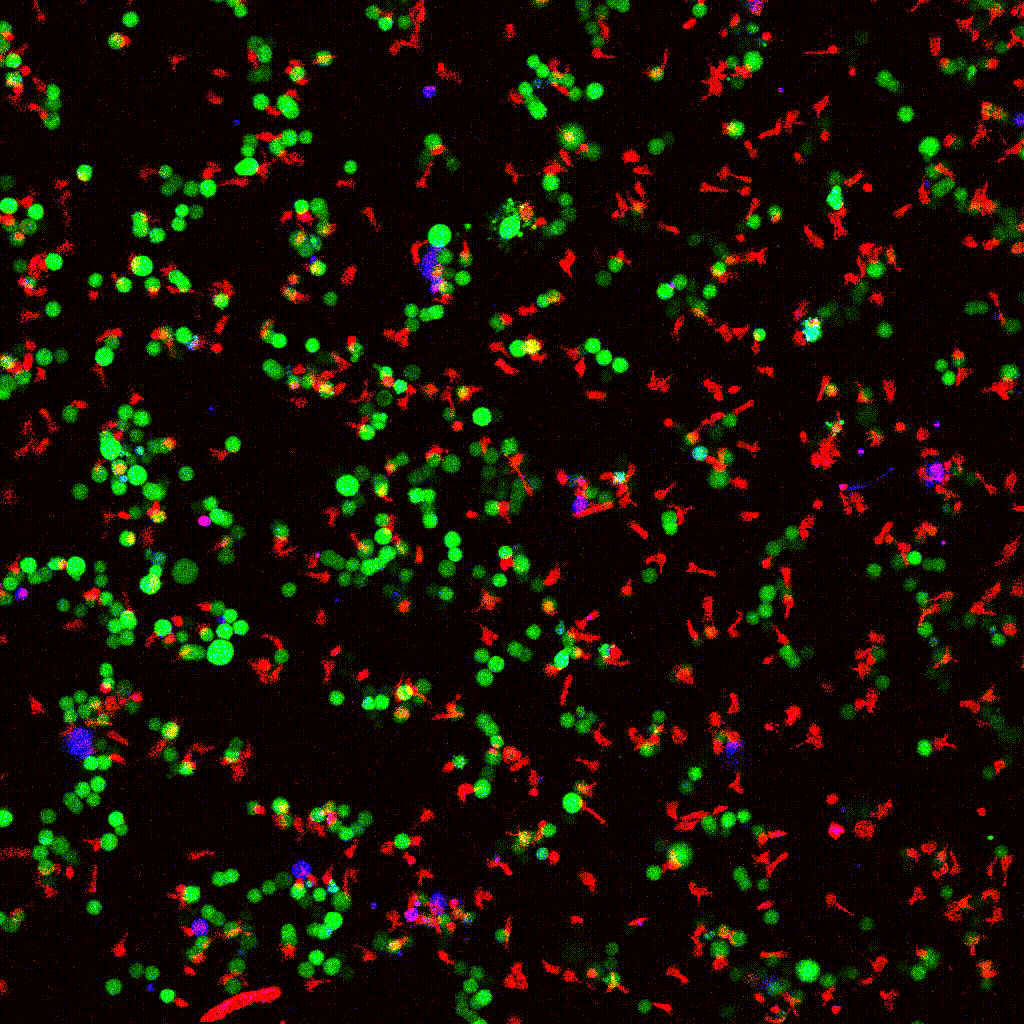
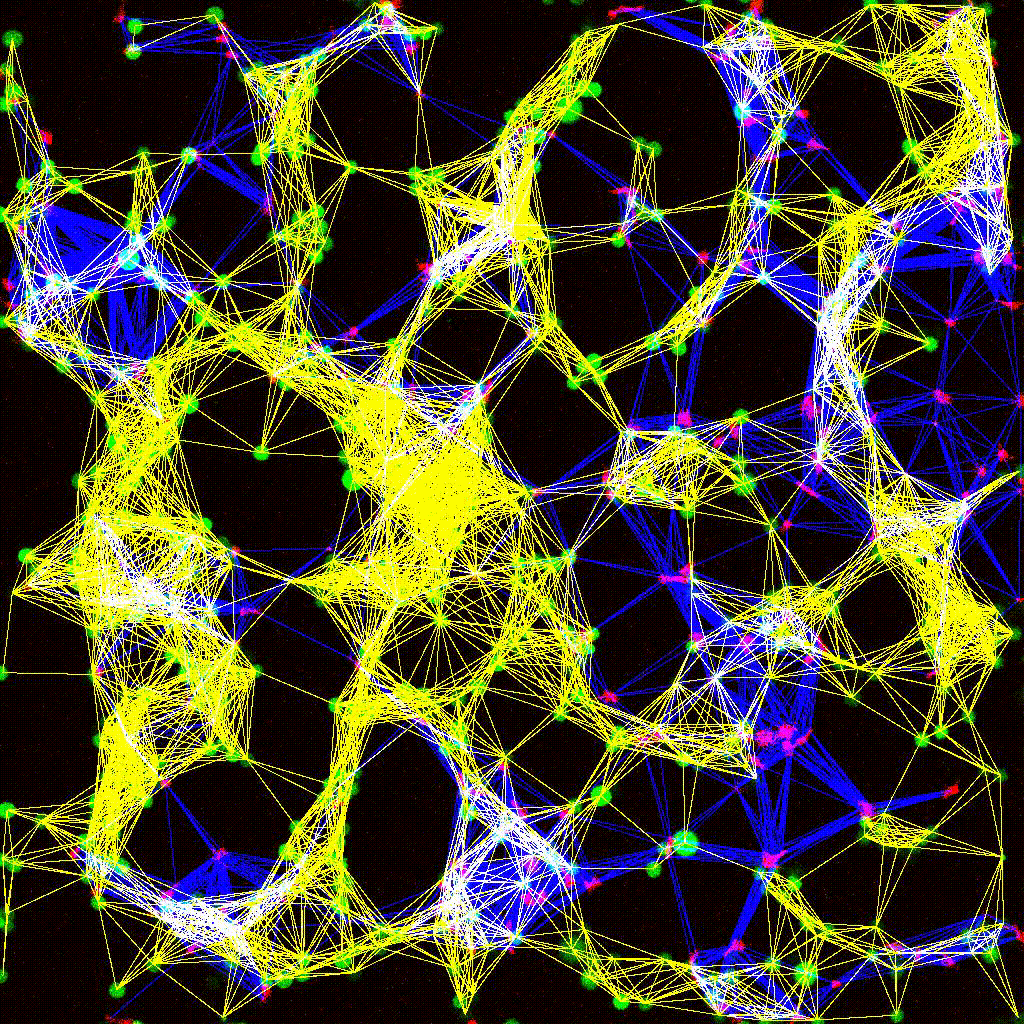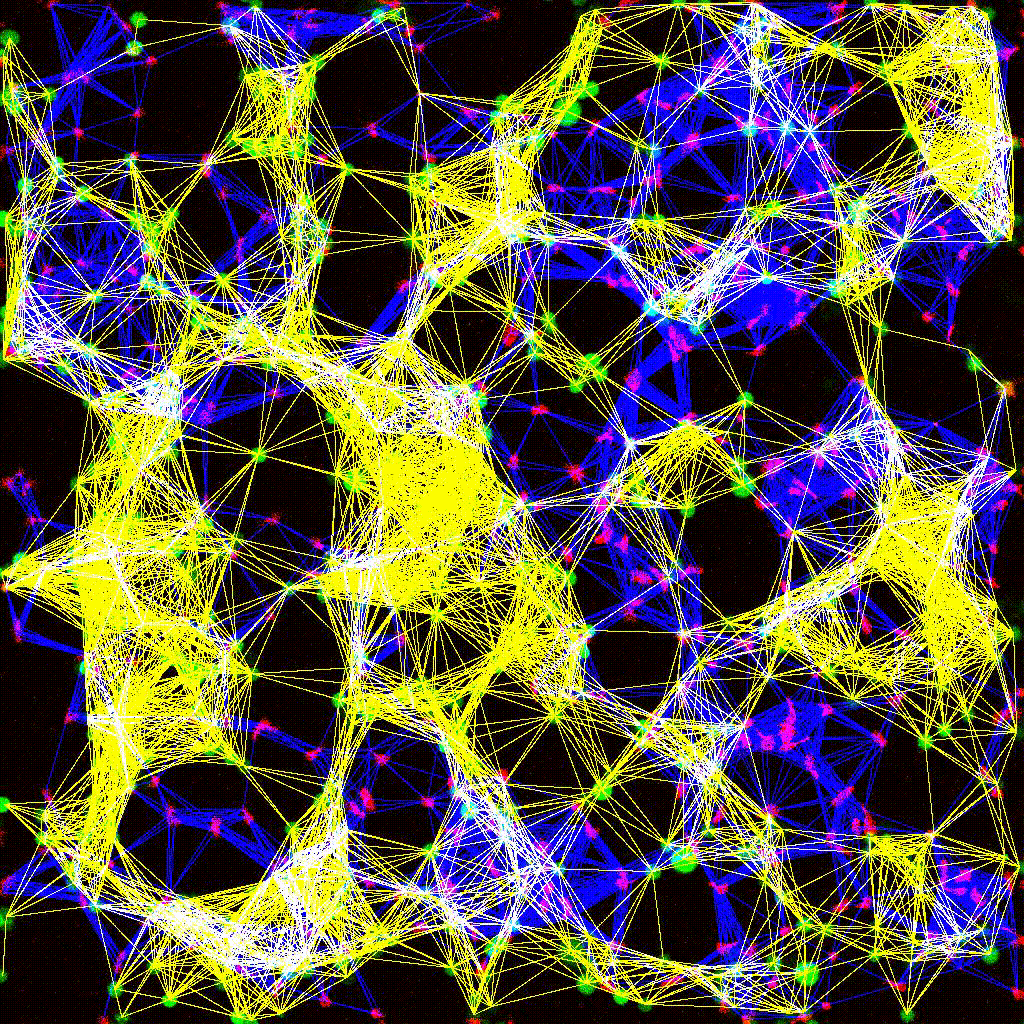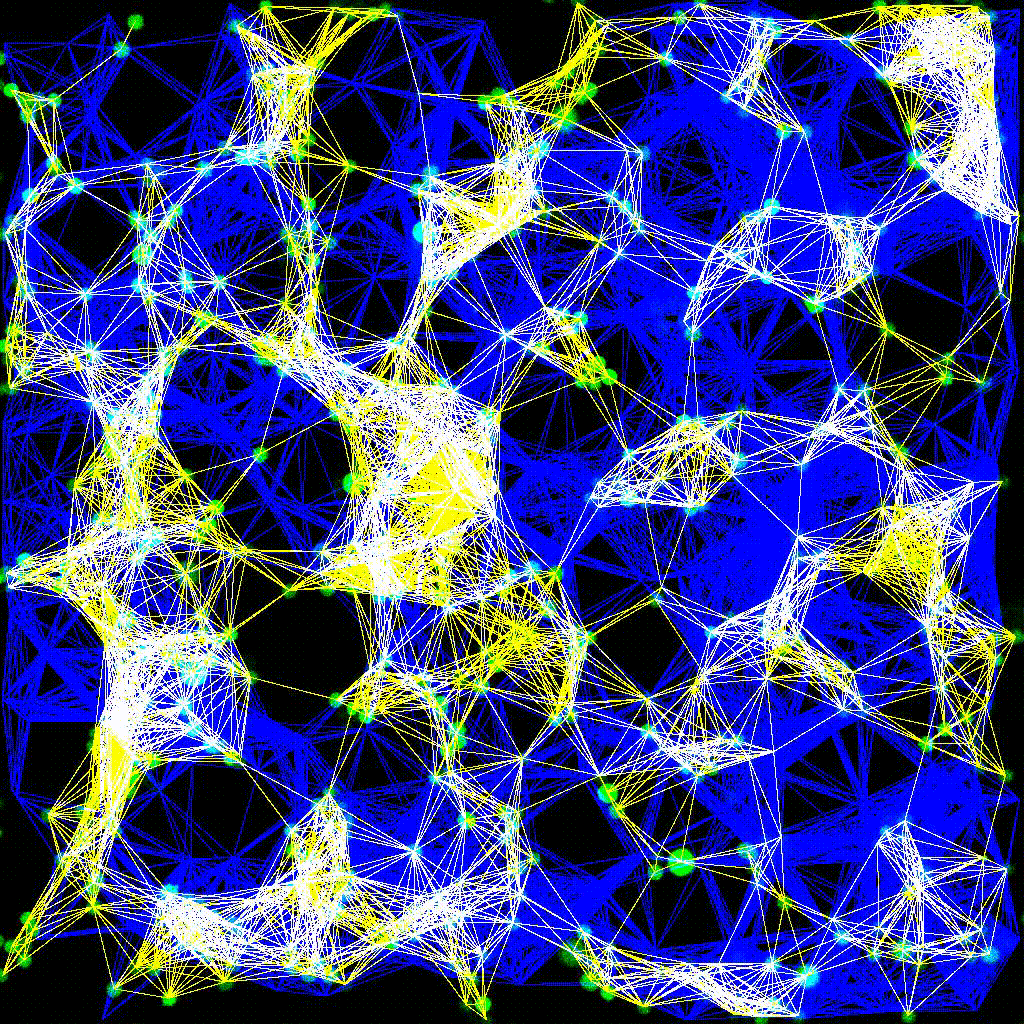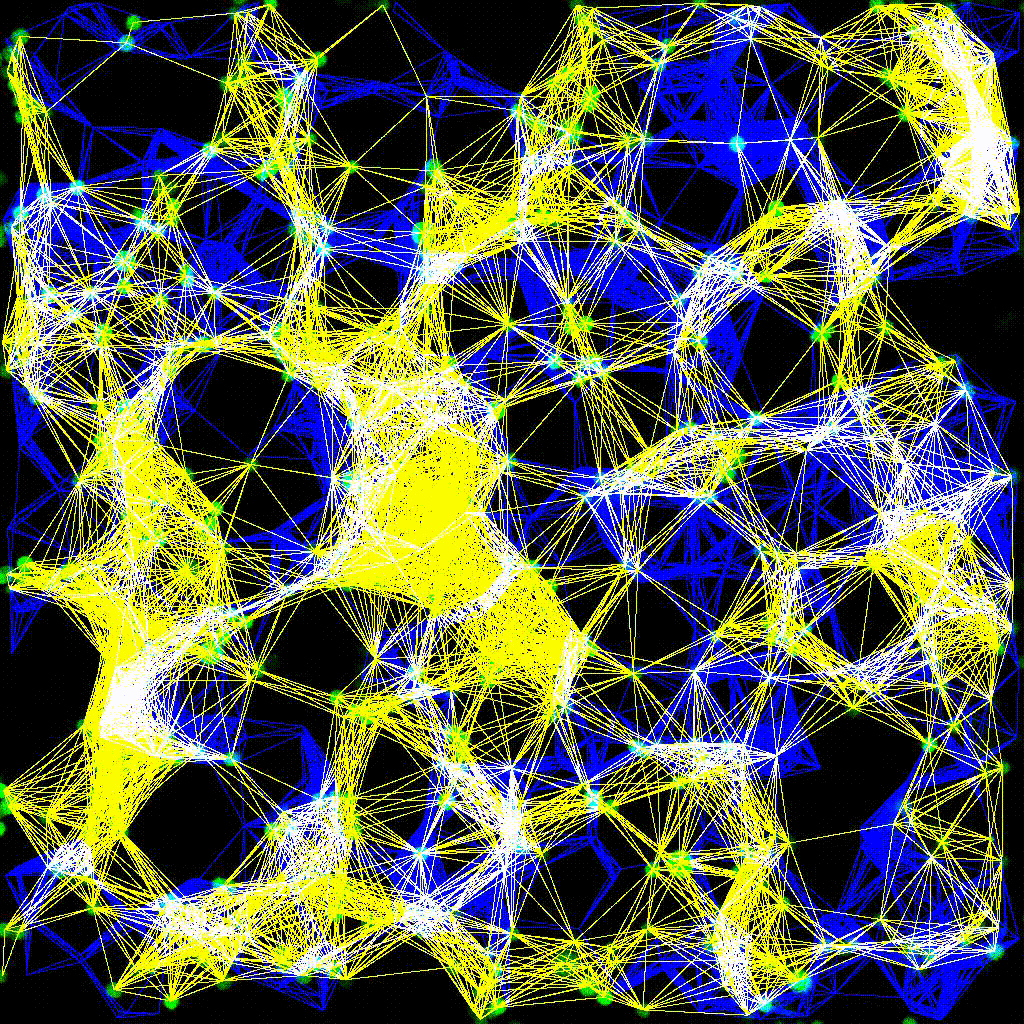
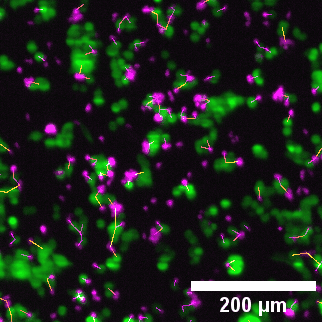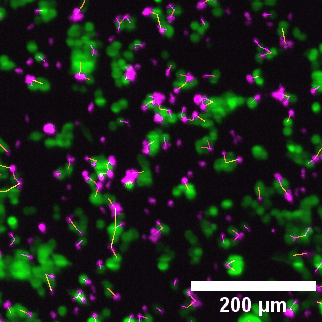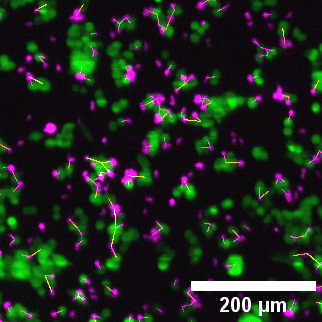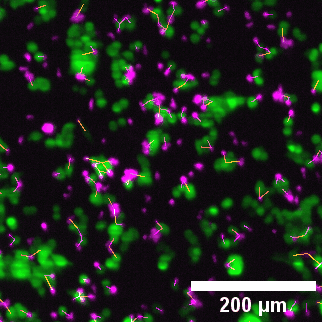
Left to right: confocal cytotoxicity assay · cell cluster networks · large-FOV lightsheet assay · zoomed contact analysis
What pyCyto Does¶
pyCyto automates the quantitative analysis of cytotoxicity experiments, where immune effector cells (e.g. T cells) kill target cancer cells under a microscope. The pipeline extracts:
Cell tracks — positions, velocities, and lifetimes over time
Cell–cell contacts — which cells touch, for how long, and with what frequency
Killing events — association between contact events and target cell death
Population statistics — survival curves, contact rates, and spatial interaction metrics
Core Concepts¶
The Compute Node Model¶
Each pipeline stage is a Python class that accepts a dictionary and returns a dictionary. This makes every stage independently testable, composable, and portable across execution environments (local, container, SLURM job):
result = MyModule(param=value)({"image": image_array})
# result["label"] → segmentation mask
Five Data Types¶
All data flowing through the pipeline has one of five types, each with a standardized dictionary key:
Type |
Key |
Description |
|---|---|---|
Image |
|
Raw or processed pixel arrays (Dask/NumPy) |
Label |
|
Segmentation masks (integer-valued arrays) |
Table |
|
Sparse per-cell feature tables (pandas) |
Network |
|
Cell interaction graphs (NetworkX) |
Arbitrary |
— |
Stage-specific outputs (plots, provenance JSON) |
Six Analysis Categories¶
Category |
Input → Output |
Examples |
|---|---|---|
Preprocessing |
image → image |
Normalization, registration, denoising |
Segmentation |
image → label |
Cellpose, StarDist |
Tabulation |
image/label → table |
Region properties, intensity features |
Tracking |
table → table |
TrackMate (via PyImageJ), trackpy |
Contact tracing |
label/table → network |
Cross-cell contact detection (pyclesperanto) |
Postprocessing |
any → arbitrary |
Survival curves, interaction plots, export |
Three Interfaces¶
pyCyto offers three ways to run pipelines — all sharing the same underlying modules:
YAML pipeline (
cyto --pipeline my_pipeline.yaml) — declarative, no Python requiredJupyter notebooks — interactive analysis and exploration (
notebooks/)SLURM batch jobs — distributed execution across HPC nodes (
distributed/)
Modular Design¶
Pipeline stages are independent classes. You can mix and match: run Cellpose segmentation locally, then submit TrackMate tracking as a SLURM job, then analyse contacts interactively in a notebook — all on the same intermediate files.
Documentation Structure¶
Section |
For whom |
|---|---|
Everyone — install pixi environments, configure Fiji |
|
Researchers — understand the full analysis flow |
|
Cluster operators — SLURM, SSH tunnels, JupyterLab |
|
Developers — add new modules, extend the pipeline |
|
Developers — PyImageJ/TrackMate integration details |
|
Operators/Developers — Docker and Apptainer usage |
|
Operators — data layout, provenance, output structure |
|
Developers — full autodoc reference |
Quick Start¶
# Install pixi (once)
curl -fsSL https://pixi.sh/install.sh | bash
# Clone and install environments
git clone git@github.com:bpi-oxford/Cytotoxicity-Pipeline.git
cd Cytotoxicity-Pipeline
pixi install
# Run the example pipeline
pixi run cyto --pipeline pipelines/pipeline.yaml -v
# Or open notebooks interactively
pixi run jupyter
See Setup for the full installation guide including GPU environments and Fiji.